Huntington Hastalığı
Günümüzde, tıp alanında ciddi manada bilimsel ve teknolojik ilerleme sağlanmış; birçok rahatsızlığa akılcı ve etkili çözümler üretilmiştir. Bununla birlikte, her ne kadar araştırma ve geliştirme anlamında başarı sağlansa da, tedavi sunma noktasında yetersiz kalınan bir takım hastalıklar hala mevcuttur. Kişileri genç yaşta ve ileri derecede etkileyen bu önemli hastalıkların başında Huntington hastalığı gelir.
Huntington Hastalığı Nedir?
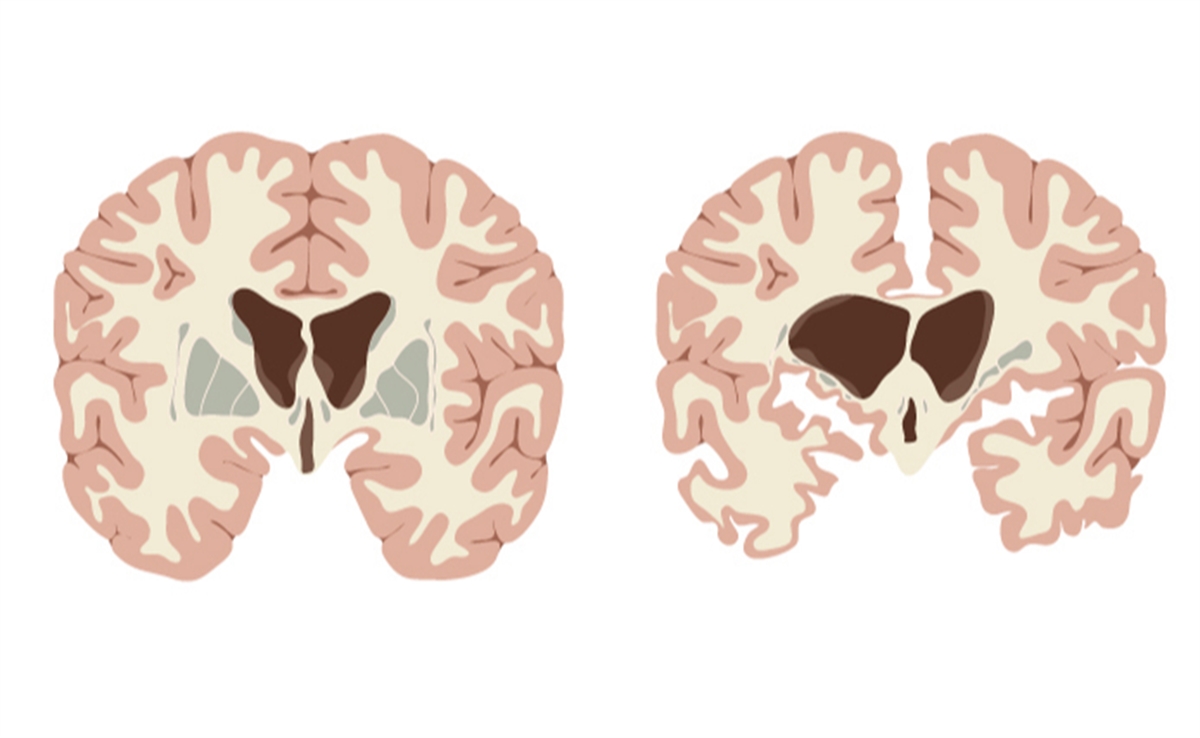
Huntington hastalığı; genetik temeli olan ve sinir sisteminin temel elemanı nöronları hedef alan, ilerleyici bir nörolojik hastalıktır. Kalıtım yoluyla ebeveynlerden çocuklara aktarılır. Ancak, gelişim süreci diğer genetik hastalıklara göre farklılık gösterir. Bu kalıtım bozukluğu sebebiyle, yaş ilerledikçe beyin dokusunda ilerleyici yıkım başlar ve bu durum genç yaşta çeşitli nörolojik belirtilerin ortaya çıkmasıyla sonuçlanır. Mevcut tıbbi koşullarda hastalığı tamamen tedavi eden bir yöntem bulunmamaktadır; tedavide, ortaya çıkan belirtilerin kontrol altına alınmasına yönelik hareket edilir.
Huntington Hastalığı Çeşitleri Nelerdir?
Huntington hastalığı; genetik alt yapıya, kalıtım şekline ve ortaya çıktığı yaşa bağlı olarak ikiye ayrılır:
- Geç Başlangıçlı Huntington Hastalığı: Huntington hastalığının en sık görülen formudur. Belirtiler sıklıkla 30 ila 40’lı yaşlarda ortaya çıkar. Kalıtımdaki bozukluk nesiller boyu hastalığın şiddetlenmesiyle orantılı olarak artar.
- Erken Başlangıçlı Huntington Hastalığı: Daha nadir görülen bu formda hastalık, sıklıkla çocukluk veya ergenlik döneminde ortaya çıkar.
Huntington Hastalığı Neden Olur?
Huntington hastalığı, insan genomundaki 4. kromozomda bulunan belirli bir gen diziliminin varlığı sonucu ortaya çıkar. Bu bölgede CAG şeklinde dizilen genetik bilginin sürekli tekrarlaması sonucu, hastalığın ortaya çıkmasına neden olan “huntingtin” isimli protein, nöronlar tarafından fazla üretilmeye ve birikmeye başlar. Biriken bu protein, nöronlarda yıkım sürecini başlatarak hastalık semptomlarının ortaya çıkmasına neden olur. Buradaki önemli nokta, genetik aktarımla sonraki nesillerde CAG dizilerinin tekrarlanmasının oranının artmasıdır. Bunun sonucu olarak, kişide CAG tekrarı ne kadar çoksa, hastalık o kadar şiddetli ve erken yaşta gelişmektedir.
Buna göre; hastalığın etkeni, 4.kromozomunda dominant (baskın) karakterde kalıtılır. Otozomal baskın karakterde olması sebebiyle hem erkekleri hem de kadınları etkileyebilir.
Huntington Hastalığının Belirtileri Nelerdir?
Hastalık belirtileri erken ve geç başlangıçlı forma göre farklılık gösterebilir.
Buna göre, geç başlangıçlı formda aşağıdaki belirtilerle karşılaşılabilir:
- Psikolojik problemler: Depresyon, psikoz atakları veya halüsinasyonlar gibi çeşitli şekillerde psikolojik sorunlar ortaya çıkabilir.
- İrritabilite: Kişiler huzursuzluk gösterebilir ve uyaranlara aşırı duyarlı olabilir.
- Koordinasyon zayıflığı: Beynin bilişsel fonksiyonları etkilendikçe günlük işleri yürütmede ve koordine etmede zorluklar başlar.
- Distoni: Vücudun çeşitli kaslarında istemsiz kasılmalar, çeşitli postür ve duruş bozuklukları, hareketleri düzgün yapmada güçlük gibi çeşitli hareket bozuklukları gelişir.
- Karar vermede zorluk: Bilişsel yetenekler etkilendikçe muhakeme yeteneği zayıflar.
- Yeni bilgileri öğrenmede güçlük: Yeni edinilen bilgilerin uzun dönem hafızaya işlenmesinde sorunlar başlar.
- Konuşma ve yutkunmada zorluk: Vücuttaki diğer kaslarla beraber, konuşma ve yutkunmadan sorumlu kaslarda da istemsiz veya düzensiz kasılmalar oluşması nedeniyle, konuşma ve yutkunma zorlaşır.
- Kişilik değişiklikleri: İleri dönemde psikolojik belirtiler şiddetlenerek kişilik değişikliklerine yol açar.
- Hafıza kaybı: Erken yaşta ciddi hafıza kaybı ve demans (bunama) görülür.
- Yürüme bozukluğu: Hareket bozuklukları, ilerlediği halde yürümenin bozulmasıyla sonuçlanır.
- İstemsiz hareketler: Düzensiz ve istemsiz şekilde, şiddetli tarzda kol ve bacaklarda hareketler izlenir.
- Erken başlangıçlı Huntington hastalığı ise aşağıdaki belirtileri beraberinde getirir:
- Tükürüğünü yutamama: Çocuklarda yutma fonksiyonunda gerileme nedeniyle tükürük yutulamaz ve ağızdan aktığı izlenir.
- Kasılma: Küçük çocuklarda belirli kaslarda kasılma ve rijidite (sertleşme) izlenir.
- Sakarlık ve sık sık düşme: Çocuklarda beklenmeyen tarzda sakarlaşma ve sık sık düşme görülür.
- Yavaş hareketler: Günlük hareketlerde azalma veya yavaş harekete geçme gözlenir.
- Konuşmada bozulma: Normalde konuşabilen çocukta gittikçe konuşmada gerileme ortaya çıkar.
- Epileptik nöbetler: Epilepsiye benzer şekilde kasılma nöbetleri gelişebilir.
- Okul başarısında ani düşüşler: Beklenmedik şekilde okul performansında ciddi azalma izlenir.
Huntington Hastalığı Tanısı Nasıl Konur?
Huntington hastalığı tanısında en önemli nokta aile öyküsünün varlığıdır. Ailede benzer şikayetlere sahip kişilerin bulunması veya tanı almış kişilerin varlığı oldukça önemlidir. Bunun yanında, kişilerin yukarıdaki belirtileri göstermesi durumunda, uzman bir hekim tarafından yapılan ayrıntılı hastalık öyküsü sorgulaması ve fiziki muayene sonrasında çeşitli tetkiklere başvurulabilir.
Bu doğrultuda, nörolojik muayene esnasında refleks muayenesi, görme ve işitme testleri, yürüyüş testleri uygulanabilir. Gerekli görüldüğünde kasların detaylı incelemesine yönelik elektromiyografi (EMG) ve beyin fonksiyonlarının değerlendirilmesi için elektroensefalografi (EEG) yöntemlerine başvurulabilir. Beyin dokusunun detaylı incelemesi için bilgisayarlı tomografi (BT) ve manyetik rezonans (MR) görüntülerine bakılır. Psikolojik değerlendirme için çeşitli psikiyatrik testlerden yararlanılır.
Son olarak hastalık tanısının kesin konulması için genetik test yapılır. Genetik test ayrıca sonraki çocuklarda hastalığın gelişme riski ve şiddeti açısından faydalı bilgiler verebilir.
Huntington Hastalığı Tedavisinde Neler Yapılır?
Mevcut tıbbi koşullarda, hastalığın kesin bir tedavisi bulunmamaktadır. Gen tedavisinde kat edilen gelişmeler hastalığın kesin tedavisinin bulunması hakkında umut verse de, klinik uygulamada herhangi bir yöntem söz konusu değildir. Bununla beraber, Huntington hastalarının yaşam kalitesini artırmak ve hayat koşullarını iyileştirmeye yönelik çeşitli yöntemler başarıyla uygulanmaktadır.
Buna göre, istemsiz kasılmaların önlenmesi ve kaslardaki sertliklerin çözülmesi için çeşitli ilaç tedavilerinden yararlanılır. Psikolojik sorunların tedavisinde ilaç tedavisi ile beraber psikoterapi faydalıdır. İlaç tedavisinin yanında, kas ile ilgili sorunların çözümünde özellikle fizik tedavi yöntemleri oldukça yararlı olmaktadır. Konuşma terapileri de ilerleyen dönemlerde ortaya çıkan konuşma bozukluklarının kontrol edilmesinde sıklıkla başvurulan yöntemlerdendir.
Tüm bu yöntemlere rağmen, hastalık ilerleyici özelliktedir ve uzun dönemde hastalığın ilerleyişini engellemek mümkün değildir. Altta yatan genetik özelliğe bağlı olarak, Huntington hastaları hastalığın gelişiminden itibaren 15 ila 20 yıl yaşayabilmektedir. Erken başlangıçlı form ise daha şiddetlidir ve gelişiminden itibaren 10 ila 15 yıl içerisinde ölüm gerçekleşebilmektedir. Ölüm sebebi olarak; hareket bozukluklarına bağlı yaralanmalar, yutma bozukluğu ile ilişkili gelişen zatürre enfeksiyonu ve psikolojik sorunlardan kaynaklı intihar girişimleri gözlemlenmiştir.